

Key Takeaways
- Profile 10 million cells from up to 1,152 samples in a single experiment
- Boost the statistical power of the analysis by increasing the number of cells assayed per sample
- Comprehensively characterize responses to perturbations or drug treatments
Experimental Design
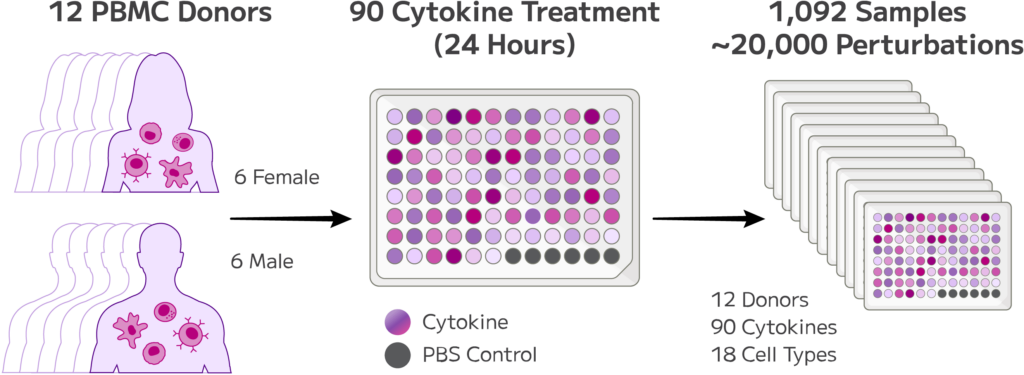
Figure 1: Overview of Experimental Design. Approximately 10 million cells from twelve healthy female and male PBMC donors treated with 90 different cytokines were processed in the GigaLab in a single experiment.
Cryopreserved PBMCs from twelve healthy donors were purchased from a commercial vendor. Samples were thawed in a 37༠C water bath, transferred to a 50 mL centrifuge tube, diluted dropwise with warm FBS media, centrifuged, and washed with cold FBS. All samples had a viability >90% after thawing.
For each donor, cells were seeded at 1 million cells per well in a 96-well plate for a total of 12 plates across all donors. Cells were treated with 90 different cytokines or PBS (control) for 24 hours resulting in a total of 1,092 experimental conditions. Next, cells were transferred into a deep-well plate, washed with PBS and centrifuged. Once supernatant was removed, cells were fixed with the Evercode Cell Fixation v3 High Throughput Plate-Based Workflow adopted for use with an Integra Assist Plus instrument. In the end, fixed samples were aliquoted into PCR plates, and stored at -80༠C. On a day prior to running the downstream Evercode whole transcriptome experiment, aliquots of fixed samples were thawed in a 37༠C water bath and counted in batches.
Fixed samples were processed with Evercode WT Mega v3. After barcoding, 62.45% of cells were retained. Sequencing libraries were made using a Hamilton liquid handler instrument. Libraries were sequenced using the Ultima Genomics sequencing platform and achieved ~31,000 mean reads per cell.
After demultiplexing, sequencing data were processed with the Parse Analysis Pipeline v1.4.0. Data were integrated with Harmony, filtered to remove low quality cells, cell types classified with Azimuth, and annotations finalized manually.
Results
A total of 9,697,974 cells across 18 immune cell types were identified (Figure 2) including rare cell types that are difficult to detect when analyzing datasets with low cell numbers. For each 1,092 experimental conditions, a median number of 7,400 cells was recovered.
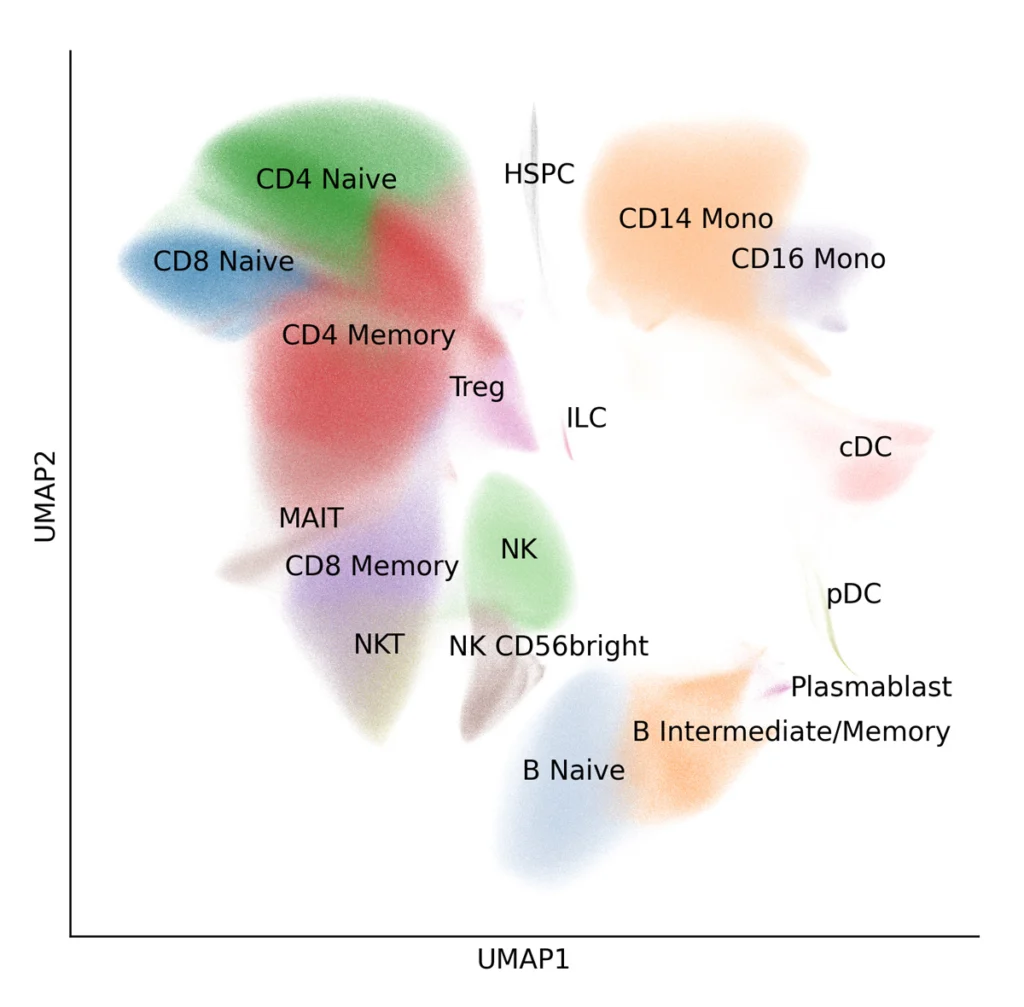
Figure 2: UMAP of 9,697,974 million PBMCs from 12 donors. Data from each donor was integrated using Harmony, clustered with Scanpy, and manually annotated.18 immune cell types containing cells from all donors and experimental conditions.
Differential expression (DE) analysis was performed to calculate the number of genes upregulated in response to cytokine treatment across all cell types (Figure 3). Many cytokines induce big transcriptional changes reflected by the upregulation of more than 50 genes.
![]()
Figure 3: Heatmap of differentially expressed gene counts identified for each cell type and cytokine treatment. Differential expression analysis was performed and the number of genes with a positive log fold change of > 0.3 and an adjusted p value of < 0.001 relative to the PBS control was calculated for every donor and cell type. The averaged differentially expressed gene counts are shown in this heatmap. Cell types are ordered by the total number of DE genes detected across all cytokines.
Example tutorial vignettes from Parse Biosciences and Fabian Theis’ lab at Helmoltz-Munich:
Parse 10M PBMC cytokines clustering tutorial
Joey Pangallo, Efi Papalexi
Parse Biosciences, Seattle, WA
Here we present an example analysis of 10 million PBMCs treated with cytokines and processed through Parse Biosciences Evercode workflow. We use the python package Scanpy. We will demonstrate steps starting from loading in the data, to preprocessing, and finally to Leiden clustering and production of UMAP plots.
Parse 10M PBMC cytokines clustering tutorial on downsampled data
Joey Pangallo, Efi Papalexi
Parse Biosciences, Seattle, WA
This tutorial describes the same workflow as the clustering tutorial above, but starts by downsampling the full dataset to 1 million cells. This can allow people to move through the tutorial more quickly and still explore a fraction of the data if CPU memory is limited.
scCODA Parse 10M PBMC cytokines
Artur Szałata, Dominik Klein, Soeren Becker, Fabian Theis
Fabian Theis lab at Helmholtz-Munich
In this tutorial, we demonstrate an analysis on cell proportion changes in 10 million PBMCs treated with cytokines. We applied scCODA (1) to various subsamples of the full 10 million cell dataset to demonstrate that measured cell proportion changes are more statistically significant when using the entirety of the dataset. This analysis illustrates the importance of performing single cell experiments at large scale when trying to determine the effects of cell perturbation experiments.
Büttner, M., Ostner, J., Müller, C.L. et al. scCODA is a Bayesian model for compositional single-cell data analysis. Nat Commun 12, 6876 (2021). https://doi.org/10.1038/s41467-021-27150-6
Parse 10M PBMC cytokines Dask workflow
Artur Szałata, Dominik Klein, Soeren Becker, Fabian Theis
Fabian Theis lab at Helmholtz-Munich
In this tutorial, we walk through basic preprocessing steps of this 10 million cell dataset using dask. Dask does not load the whole AnnData object into memory. Instead, it loads it chunk-wise, thus requiring less CPU memory to process the dataset. We demonstrate the steps through highly variable gene selection, which is usually the step before the AnnData object can be subset placing a lesser burden on CPU memory.
We're your partners in single cell
Reach out for a quote or for help planning your next experiment.