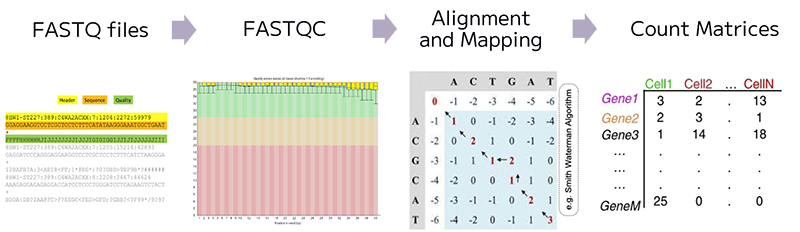
Quality control involves filtering out unwanted data, identifying artifacts like dead cells and doublets, and addressing contamination issues to ensure that only high-quality data are retained for downstream analysis.
Some of the most important pitfalls to watch out for in the data and should be included as a standard part of the downstream analysis are the following:
Removing Background or Empty Droplets
To understand single cell quality control, we must first understand what to expect in an ideal scenario.
For example, in combinatorial barcoding methods each cell is expected to be uniquely barcoded with a low percentage of doublets and minimal free-floating mRNA that could be mistakenly barcoded and retained. In droplet-based technologies, each cell should be encapsulated within a distinct oil droplet, with as few free-floating transcripts as possible.
In less ideal scenarios, multiple cells within a single droplet or barcoded together, as well as droplets containing free-floating transcripts, with or without cells result in barcoded background RNA. Combinatorial barcoding is less susceptible to background issues due to separate in-cell barcoding (Figure 2).
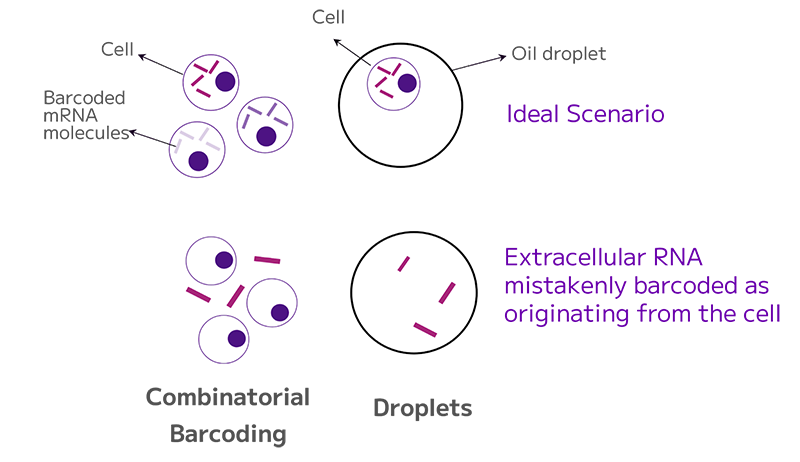
Figure 2: Only the cell’s barcoded RNA should be retained. When free floating RNA is barcoded, it results in background RNA.
To prevent the issue, starting with a cell suspension with minimal debris or damaged cells greatly improves the downstream analysis, saving time during data processing. For a more in-depth discussion on this topic, refer to our previous article on sample preparation.
But if background RNA is present, it can be computationally removed.
One method for filtering data is the classifier filter, which employs a mathematical model to estimate the composition of the dataset. It identifies barcodes that correspond to real cells as opposed to background noise.
A user-defined false discovery rate (FDR) threshold removes barcodes with a high probability of being empty. This method is particularly effective for droplet-based datasets, where distinguishing between real cells and background is crucial.
Tools like knee plots are also helpful for distinguishing biological cells from background.
Knee plot filters allow researchers to apply a hard threshold based on the inflection point visible in the curve, helping to distinguish biological cells from barcodes associated with background.
A good starting point is to set a transcript threshold, such as 200 or 500 transcripts per cell at standard read depth and adjust this based on the biological context and characteristics of the samples.
Identifying Dead or Dying Cells
Another key step in QC is identifying dead or dying cells. As cell membranes become fragile, transcripts begin to leak out, resulting in a lower overall transcript count in the cytoplasm. However, mitochondrial transcripts remain intact within the mitochondria as it is membrane bound. This can lead to a higher fraction of mitochondrial reads after cell lysis and sequencing.
There are two filtering approaches. One is setting a hard threshold based on biology. The second uses the distribution of the mitochondrial read fractions in the dataset.
Mitochondrial read fractions can be utilized to plot the percentage of mitochondrial reads relative to the number of cells or the total transcripts per cell to set appropriate thresholds.
A commonly used threshold is 10–20%. However, this may vary based on cell type – stressed cells may require a higher threshold to avoid excluding important data points, whereas nuclei should have no mitochondrial reads, as mitochondria are absent.
Identifying and Removing Doublets
Doublets occur when two or more cells share the same barcode, which can introduce artifacts in the data. Doublets can arise in both droplet-based and combinatorial barcoding methods, but they are more common in droplet-based technologies due to factors like flow rate adjustments and cell loading density (Figure 3).
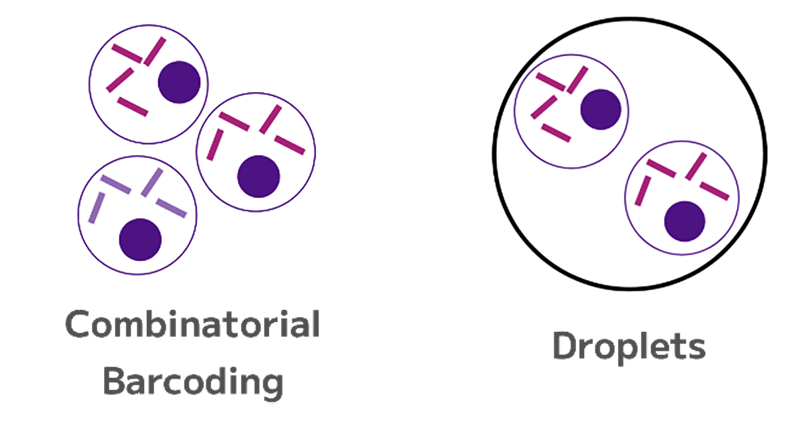
Figure 3: In a doublet, two or more cells have picked up the same barcode or barcode combination.
Several tools are available to identify and remove doublets bioinformatically—Scrublet for Python users, SC DoubletFinder for R users—that have shown strong performance in benchmarking studies.
These tools generate artificial doublet profiles and use scoring systems to assess the likelihood that a cell’s expression profile matches that of a doublet. A key input for these tools is the expected doublet rate, which depends on the chosen methodology and the characteristics of the sample.
Red Blood Cells (RBCs) and Ambient RNA
Contamination is another common challenge, such as the presence of red blood cells (RBCs) in PBMC datasets.
A cluster characterized by many transcripts per cell but few genes per cell may indicate RBC contamination. To remove them, dimensionality reduction techniques like UMAP makes it easier to exclude them from the analysis.
Ambient RNA refers to free-floating transcripts that are inadvertently barcoded alongside intact cells, a problem often seen in droplet-based technologies due to their compartmentalization methods. Tools like SoupX, CellBender, and DecontX can help mitigate ambient RNA contamination and also provide guidance on when to apply them during preprocessing to effectively remove unwanted RNA.
Remove Batch Effect
Batch effects are a common issue in single-cell datasets, as samples are often processed under different conditions, with varying technical factors such as handling personnel, reagents, or even different technologies. These systematic differences can obscure genuine biological variations.
It is advisable to remove these technical differences if they appear on UMAP plots. Data integration is the solution for removing such batch effects.
For data integration and batch effect removal, the appropriate tool will depend on factors like sample variation. For instance, time-course studies with different cell types at each time point need a different tool than studies with consistent cell types across samples.
Tools to consider include Seurat, SCTransform, FastMNN, scVI, and others.