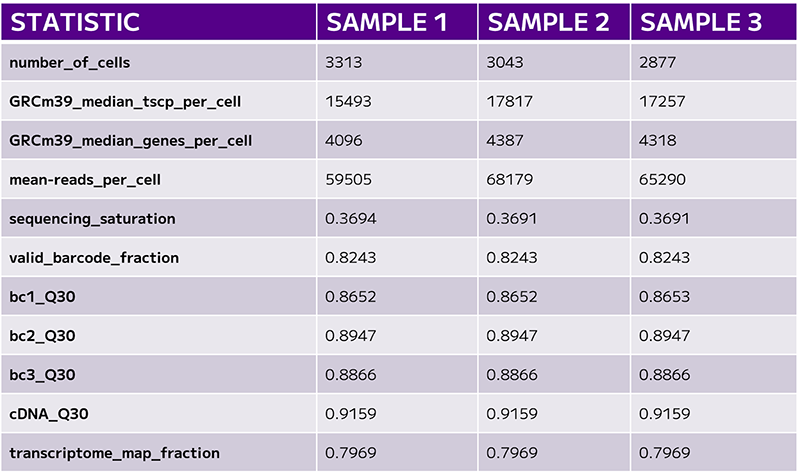
One of the first key metrics to consider in single cell sequencing experiments is the total number of cells recovered. This value is typically defined by the barcode rank plot, and it’s essential to compare it against the user’s expectations. For example, what was the target number of cells for sequencing, and how many were actually recovered? Accurate cell counting is crucial.
The next set of quality metrics includes median transcripts, median genes, and reads per cell.
The median transcript count reflects the unique transcripts used to identify gene expression, which in turn correlates with the quality of the cells in the assay.
However, the median gene count is often one of the more critical metrics when evaluating sequencing quality. This value should be considered alongside sequencing saturation—the proportion of the library that has been sequenced to its full depth. If sequencing saturation is low, it might explain a lower gene detection rate, though this could also result from working with a sample type that naturally has fewer transcripts. Setting appropriate expectations for the experiment and confirming whether the data fall within an acceptable range is vital for interpreting the results.
Reaching the targeted reads per cell is another important consideration as it impacts the sequencing saturation.
Sequencing saturation is a valuable measure of how much capacity remains for additional gene detection in a sample.
Depending on the population in focus, different saturation levels may be adequate: if the focus is a rare cell population, then higher saturation may be required to capture less abundant cell types.
However, there should be a balance between the cost of sequencing, the sequencing depth required to answer the question, and the saturation level.
Evaluating Q30 Scores and Transcriptome Map Fraction in Sequencing Quality Control
The final quality control check in sequencing analysis focuses on Q30 scores, which are crucial for evaluating the overall accuracy of your sequencing run. Q30 scores relate to per-base quality scores and indicate the percentage of reads with a Phred score of 30 or higher, meaning a 99.9% base call accuracy.
A Q30 score in the range of 80% or higher is considered excellent, indicating high-quality sequencing. However, if lower Q30 scores are observed, it could suggest problems with the sequencing run that could lead to inaccurate conclusions. A consistently low Q30 score might indicate a failed run, prompting the need for further investigation.
Should that be the case, and if a FastQC report has not yet been generated, it would be advisable to run this analysis on the FASTQ files to identify potential causes for the lower quality, particularly with respect to barcode performance.
Another critical metric to assess is the transcriptome map fraction, which measures the proportion of reads with valid barcodes that align with annotated genes in your genome of interest. This is an important indicator of how well the sequencing data reflects the actual biological content of the sample (Table 1).
Table 1: The final set of QC evaluates cell numbers, gene counts, and sequencing saturation.
The Importance of Tertiary Analysis in Single-Cell Sequencing Workflows
While secondary analysis helps assess the quality of your sequencing data, tertiary data analysis begins to interpret and extract meaningful insights from the results. This stage allows us to ask scientific questions, and dive into functional analysis. Tertiary analysis involves the use of tools like Seurat, Scanpy, or platform-specific methods such as Split-Pipe and its web-based cloud version, Trailmaker.
This phase is the most time-consuming part of the bioinformatics workflow, as it requires iterative analysis, refinement, and ultimately experimental validation. It will require multiple rounds of analysis to hone in on the findings that are most relevant to the study. Though this process demands time and patience, it is also where the true value of your data is realized. We will explore this in more detail in the next article.


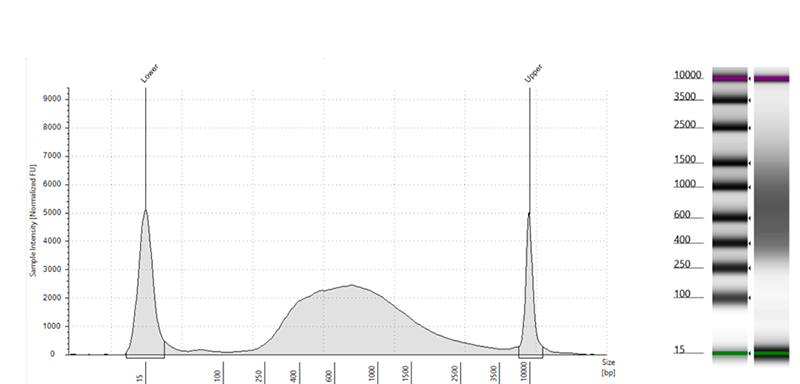
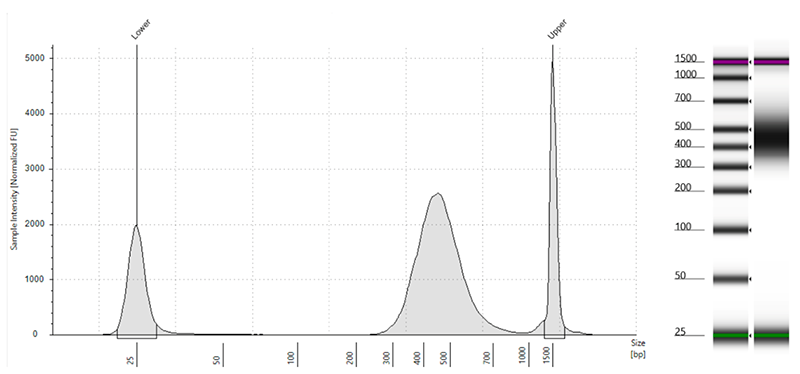
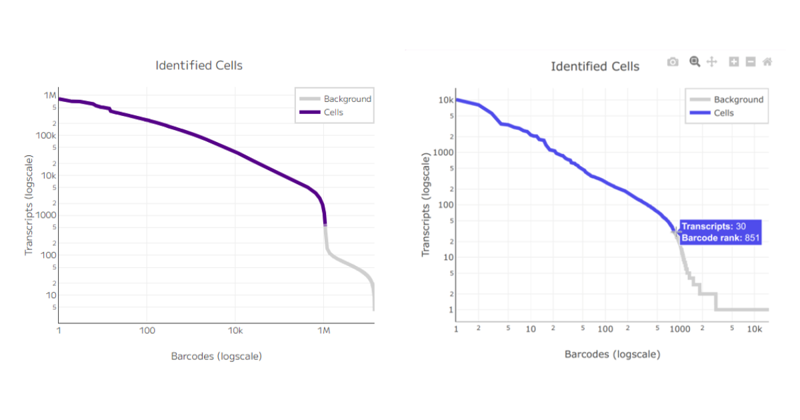 Figure 4: A cell cutoff threshold with a distinct inflection point in the curve indicates a successful experiment (panel on the left) whereas a smooth line indicates an excess of dead and damaged cells (panel on the right).
Figure 4: A cell cutoff threshold with a distinct inflection point in the curve indicates a successful experiment (panel on the left) whereas a smooth line indicates an excess of dead and damaged cells (panel on the right).