Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
BLOG › Customer Profiles › Exploring the Frontiers of Prion Research: A Q&A with Emmanuelle Vire, Ph.D.
Exploring the Frontiers of Prion Research: A Q&A with Emmanuelle Vire, Ph.D.
August 17, 2023
|
11 min read
Updated:May 24, 2024
Creutzfeldt-Jakob disease (CJD) took center stage in the mid-1990s as a new variant, the Mad Cow Disease, wreaked havoc among cattle and humans in Great Britain.
The variant – now identified as vCJD – is transmitted from cattle to humans ingesting infected meat.
The epidemic emphasized the necessity among biologists to confront interspecies disease transmission as an emergent public health risk. The UK, where the cattle to human transmission was initially identified, most closely experienced the threat. The epidemic led to the death of many.
CJD, a prion disease, is a rare, invariably fatal, and rapidly progressive neurodegenerative disorder. It is caused by a misfolded protein that replicates and accumulates in the brain. Such accumulation leads to severe neuronal loss, memory impairment, personality changes, difficulty moving, and death. CJD symptoms present like other neurodegenerative diseases, and clinicians initially find it challenging to discriminate between them.
Currently, there is no treatment for CJD.
Dr. Emmanuelle Vire is an associate professor at University College London (UCL). There, she led a team of scientists studying what happens in the brain of patients with prion diseases using cutting-edge technologies, including single cell RNA sequencing (scRNA-Seq).
Dr. Vire brought her prior expertise in epigenetics to study CJD – a disease caused by proteins – and integrate information her teams gather from genetic, epigenetic, and protein standpoints.
We recently sat down with Dr. Vire to discuss the current state of her research, particularly CJD.
Can you tell us about your scientific background and what sparked your interest in epigenetics?
I grew up in Belgium, where I studied biomedical sciences. I was lucky to do my Ph.D. in Dr. Francois Fuks’s lab, working on epigenetics. In his lab, I found a great mentor, a fantastic team, and a successful career start.
After my Ph.D., I moved to the United Kingdom to pursue postdoctoral work in Tony Kouzarides’ lab at The Gurdon Institute at the University of Cambridge. Tony is a pioneer in epigenetics and a wonderful mentor who cares for his people. Most of the epigenetic research in his lab then focused on cancer, so I worked on mechanisms underlying epigenetic modifications and their impact on cancer.
In 2015 I joined the MRC Prion Unit at the University College in London. They needed someone with epigenetic expertise to investigate the relationship between DNA, RNA, environment, and prion diseases. I worked with Prof. Simon Mead in a multidisciplinary team of clinicians, protein biologists, and geneticists.
Prions are fascinating for many reasons, mainly because — for reasons that are still unclear — they are infectious.
“Prions are fascinating for many reasons, mainly because — for reasons that are still unclear — they are infectious.”
Dr. Emmanuelle Vire
What is prion disease?
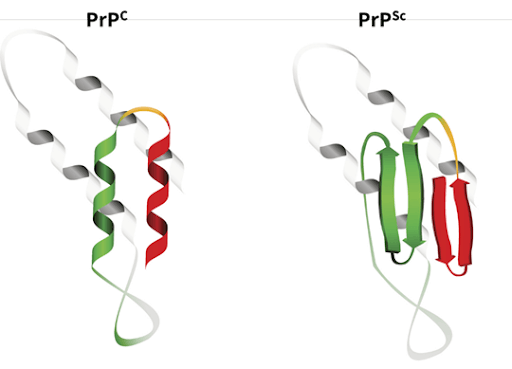
Prion disease is a type of neurodegeneration caused by a misfolded protein. Prions are proteins that can change shape, accumulate and become toxic to the brain when they do so. These proteins have two “behaviors”: one regular and one harmful. The harmful prions stick together and form aggregates that the brain cannot eliminate (Fig 1).
Fig1: PrPC is a normal protein found on cell membranes. PrPSc, or prion, is the proteinase-resistant form and can convert normal PrPC proteins into infectious isoforms by changing their conformation. This, in turn, alters the way the proteins interconnect. PrPSc causes prion disease.
Why did you choose prion disease as a model?
We use prion disease as a model for several reasons. First, it progresses much faster than other neurodegenerative diseases, which makes studying them more manageable. Further, it is infectious – it can be transmitted without modifying the genetic background. This allows us to explore it in its “natural” setting.
However, there are also some challenges. Because prion disease is infectious, we must follow strict safety procedures to protect our researchers and the general public. We require biosafety level three (BSL3) facilities, which are expensive and need highly trained staff. It’s also hard to find collaborators who are willing to work with prion samples. Finally, because prion disease is rare, funding is scarce.
But prion disease is a significant model. There is still much to learn from prion research. We want to expand the scope of research beyond neurodegeneration.
“Prion disease is a significant model. There is still much to learn from prion research. We want to expand the scope of research beyond neurodegeneration.”
Dr. Emmanuelle Vire
What purpose do prions serve?
Prions are found in yeasts and some mammals but affect these species differently.
In yeasts, prions can be beneficial and help them adapt to stressful conditions, such as an acidic environment or the presence of antifungal drugs. To our knowledge, in mammals, misfolded prions are harmful.
Not all prions have this ability to cross species barriers. Prions come in strains, just like viruses do. But unlike viruses, prion strains seem to be solely determined by their structure, not by the genetic material they carry.
“Prions come in strains, just like viruses do. But unlike viruses, prion strains seem to be solely determined by their structure, not by the genetic material they carry.”
Dr. Emmanuelle Vire
Can you briefly overview your study on diagnosing Creutzfeldt-Jakob disease using miRNA signatures?
I was interested in microRNAs because they are stable and present in biological fluids. It was a longitudinal study.
We collected blood samples from patients before they developed any symptoms of neurodegenerative diseases. We knew the time when they started showing signs of cognitive decline. It’s not a fixed date on the calendar but a time when their condition changed.
We found that by analyzing the expression of a specific pattern of three microRNAs (miRNAs) in the blood sample, we could differentiate between patients who had Alzheimer’s disease and those who had sporadic Creutzfeldt-Jakob disease (CJD) even before they became symptomatic.
How close are we to a non-invasive diagnostic test for CJD?
Currently, the only definitive way to diagnose CJD is by examining the brain tissue after death. CJD diagnosis relies on ruling out other possible causes, but confirmation can only come with an autopsy.
Our earlier work showed that a simple blood test in the research lab could help us to distinguish between Alzheimer’s and sporadic CJD cases, regardless of gender and age. However, the test is based on three miRNAs, and turning this into a clinical test is difficult.
What are the challenges involved with converting this into a clinical test?
Measuring the expression of miRNAs in a biological fluid is challenging, especially for rare diseases with elusive biomarkers. Therefore, a three-miRNA signature may be insufficient and may need to be combined with other markers or imaging techniques.
But we should keep pursuing the quest for non-invasive lab tests. They would provide definite answers much faster — all stakeholders, patients, families, and clinicians need these.
What can you tell us about the gene expression changes observed in your prion research?
We used a well-established model of prion disease to study the longitudinal changes in gene expression as the disease progresses. We carefully designed the experiment to remove confounding factors such as age, gender, batch effects, and other biases.
We used a single-cell approach to identify which cell types in the brain were affected by the disease and when.
Our goal was to identify the earliest cellular targets of the disease, the timing and nature of their gene expression alterations, and the link between these changes and the clinical manifestations. We also validated our model results by examining how well they matched with human brain tissue from deceased patients.
We used Evercode WT for single-cell sequencing. We chose the tissue and time points based on the infectivity curve, which measures how many infectious particles are in the tissue.
“We used a single-cell approach to identify which cell types in the brain were affected by the disease and when.”
Dr. Emmanuelle Vire
The infectivity curve has two phases: an exponential phase and a plateau phase. The exponential phase is when the infectivity increases rapidly but strikingly with almost no symptoms. The plateau phase is when the infectivity level stabilizes (no exponential increase anymore), but the brain shows signs of damage and dysfunction.
One of our most surprising findings was that there was close to no change in gene expression during the exponential phase.
Such findings are truly remarkable.
What are the implications of this finding?
Our results are consistent with the protein-only hypothesis, which states that the conformational change of a native protein causes prion disease.
The gene expression changes we observed as the disease progressed were mainly related to known transcripts in the neurodegeneration field. For example, we saw the differential expression of the glial fibrillary acidic protein (GFAP), an astrocyte activation marker.
We confirmed this increase with RNAscope, and it was very striking.
We also know from other studies that GFAP is elevated in sporadic CJD patients’ blood and cerebrospinal fluid, but that effect is not specific to CJD. Indeed, increased GFAP is also a marker of Alzheimer’s Disease.
So our data suggest similar pathways may be shared in Alzheimer’s and CJD brain cells.
How did Evercode help with the logistics of the experimental study?
It’s the reason we chose it. We wanted a protocol we could use in-house.
I want to acknowledge the fantastic work and perseverance of the Ph.D. student who led this work, Athanasios (Thanos) Dimitriadis. He was the driving force behind the whole project. He adapted the Evercode protocol to make it compatible with BSL3 conditions, working with prion-infected samples.
“Athanasios (Thanos) Dimitriadis was the driving force behind the whole project. He adapted the Evercode protocol to make it compatible with BSL3 conditions, working with prion-infected samples”
Dr. Emmanuelle Vire
Thanos had to overcome many challenges and make adjustments, but he never gave up. He efficiently collaborated with our institute’s safety committee and experts to ensure the protocol was safe to use with prions.
Later, when Parse came out with their improved version of Evercode, we compared it with the original method. We found that Parse improved our capture efficiency and allowed us to see more of the transcriptome.
We also appreciated the customer service and troubleshooting support from Parse. They were very accommodating and responsive.
“We found that Parse improved our capture efficiency and allowed us to see more of the transcriptome.”
Dr. Emmanuelle Vire
What aspects of working with Parse technology were most helpful to your work?
I mean, very pragmatically, it was the recommendation of the Parse support team to include the sucrose gradient in the nuclear suspension preparation. That made a massive difference for us.
We also appreciated the customer service and the troubleshooting live with the experts. With the help of Parse, we improved the quality of the cell suspension and removed hurdles without too much sweat and tears.
What advice would you give to other scientists pursuing single cell sequencing studies in prion disease?
I would like to see how different strains of prions affect the transcriptome of different tissues. We only looked at one model of sporadic CJD, but many other models reflect the diversity of human diseases. Someone should build an atlas of prion diseases and their effect on tissues/cells.
We need more collaboration among researchers worldwide because these are rare diseases, and funding is scarce. Instead of competing, we should share more and work together. Patients and their families are expecting us to do so. We owe them that! I wish the prion community could lead the way in building a collaborative model for research on rare diseases and welcome more integration across disciplines.
There is room for a consortium on single cell in prion diseases, or even better, one that addresses some of the critical questions we need to answer for the patients. We have a responsibility for using the samples they donate to the research community as best as possible.
We have the technology, the talent, the motivation, and the questions. What’s stopping us? Let’s collaborate more and break down the barriers to progress.
“We have the technology, the talent, the motivation, and the questions. What’s stopping us? Let’s collaborate more and break down the barriers to progress.”
Dr. Emmanuelle Vire
Thank you, Dr. Vire, for your availability to talk with us and share your knowledge about prion diseases. If interested, please find more of Dr. Vire’s work on CJD below.
Drs. Dimitriadis and Vire’s latest work identifying dynamic gene signatures within the single nuclei of mammalian prion diseases is currently in the peer review process and is anticipated to be published soon.
Viré EA, Mead S. Gene expression and epigenetic markers of prion diseases. Cell Tissue Res. 2023;392(1):285-294. doi:10.1007/s00441-022-03603-2
Kroll F, Dimitriadis A, Campbell T, et al. Prion protein gene mutation detection using long-read Nanopore sequencing. Sci Rep. 2022;12(1):8284. Published 2022 May 18. doi:10.1038/s41598-022-12130-7
Guntoro F, Viré E, Giordani C, et al. DNA methylation analysis of archival lymphoreticular tissues in Creutzfeldt-Jakob disease. Acta Neuropathol. 2022;144(4):785-787. doi:10.1007/s00401-022-02481-w
Dabin LC, Guntoro F, Campbell T, et al. Altered DNA methylation profiles in blood from patients with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. 2020;140(6):863-879. doi:10.1007/s00401-020-02224-9
About the Author
Laura Tabellini Pierre
Laura Tabellini Pierre, MSc, is a scientific and technical writer at Parse Biosciences with extensive experience in immunology, encompassing both academic and R&D research.