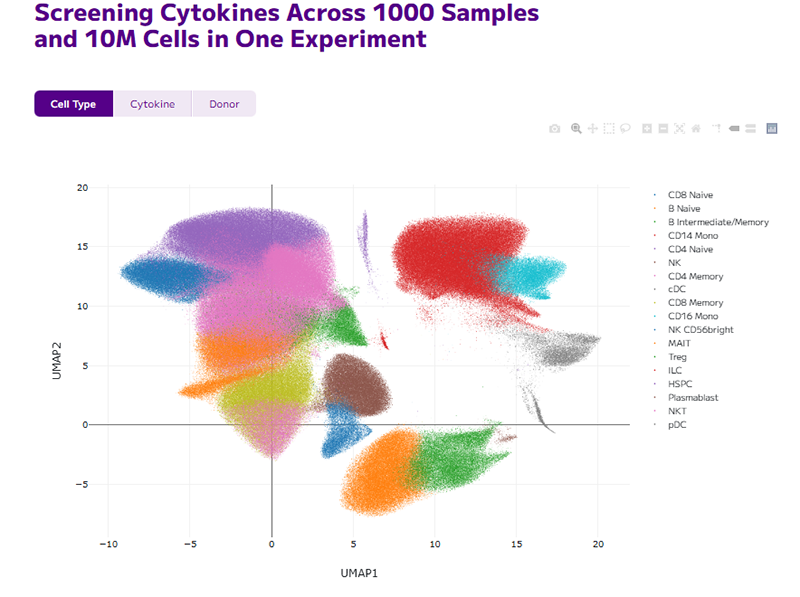
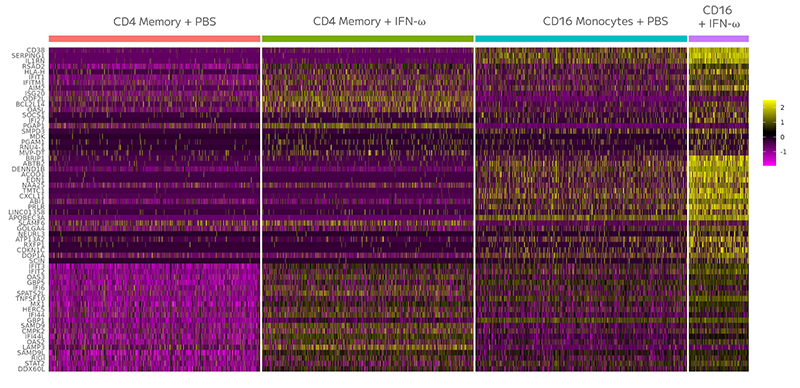
Using scRNA-seq researchers can assess the response of various cell populations in tissue samples to fine-tune drug dosage and enhance safety before clinical trials.
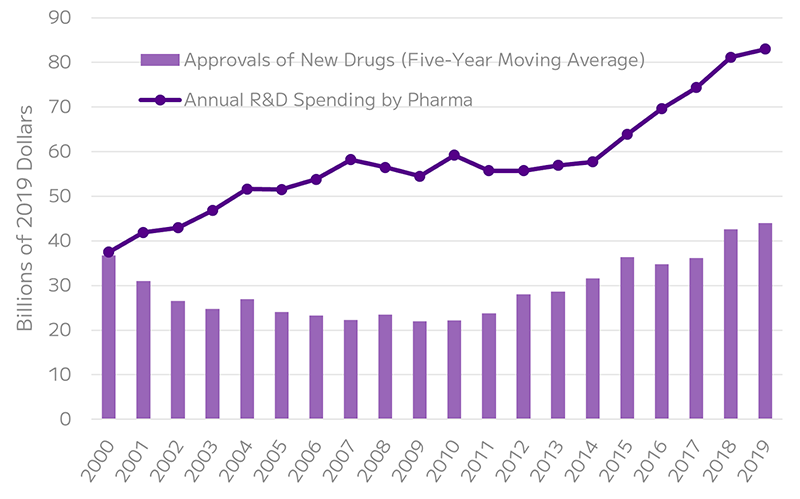
By predicting pharmacokinetics and potential toxicity early in the drug discovery phase, scRNA-seq can help filter out likely failures, reducing costs and improving success rates by minimizing investments in unsuccessful candidates. This approach is vital in addressing the high attrition rates often seen in clinical trials.
Target Identification and Validation
RNA sequencing at the single cell resolution is crucial for identifying genes linked to specific cell types or novel states involved in disease, aiding in the discovery of potential drug targets. For target validation, scRNA-seq supports gathering evidence across disease biology and target tractability. Study models, including cell lines and patient-derived organoids, can be evaluated to reveal cell-type-specific transcriptomic responses and pathway alterations related to disease states.
When scRNA-seq is used to analyze CRISPR perturbations, detecting the target genes and the cascade of pathway modifications triggered helps researchers understand complex interactions within cellular networks. This approach provides insights into gene function, regulatory mechanisms, and potential therapeutic targets.
Combining scRNA-seq with CRISPR screening allows for the large-scale mapping of how regulatory elements and transcription start sites impact gene expression in individual cells. This approach has been applied to profile approximately 250,000 primary CD4+ T cells, enabling systematic mapping of regulatory element-to-gene interactions and the functional interrogation of non-coding regulatory elements at the single-cell level.
Drug Screening
Traditional drug screening relies on general readouts like cell viability or marker expression, lacking comprehensive detail. ScRNA-seq enables detailed cell-type-specific gene expression profiles, essential for understanding drug mechanisms.
High-throughput screening now incorporates scRNA-seq for multi-dose, multiple experimental conditions, and perturbation analyses, providing richer data that support comprehensive insights into cellular responses, pathway dynamics, and potential therapeutic targets.
This approach enables researchers to identify subtle changes in gene expression and cellular heterogeneity, enhancing the understanding of drug efficacy and resistance mechanisms.
Biomarkers Identification and Patient Stratification
Biomarkers are characteristic features of a process that can be objectively and reproducibly assessed and measured. They can be prognostic, diagnostic, predictive, monitoring biomarkers.
Traditionally, biomarkers have been identified using several techniques. The advent of RNA sequencing has significantly accelerated this process, but bulk transcriptomics, while historically used to identify cancer biomarkers, fails to capture cell population complexity.
ScRNA-seq has advanced this field by defining more accurate biomarkers, such as those in colorectal cancer, leading to new classifications with subtypes distinguished by unique signaling pathways, mutation profiles, and transcriptional programs.
This deeper molecular understanding enables to evaluate the risk of developing a disease, monitor a disease course, and make an accurate diagnosis. It allows for more precise stratification of patients, tailored therapeutic strategies, and improved predictions of treatment responses, ultimately contributing to better clinical outcomes and personalized medicine approaches.